
Is dietary cholesterol required for good human health?
A comprehensive guide on dietary cholesterol


This article is a collaboration between Dr. Mike Hunter and Kat Fu, M.S., M.S. (The Longevity Vault). Dr. Mike Hunter is a retired physician specializing in Internal Medicine and Gastroenterology, with over 20 years in academic medicine. Dr. Hunter has headed medical units and the Gastroenterology Department at Helen Joseph Hospital, University of the Witwatersrand, South Africa, and practiced in the NHS in the UK. I have lectured at all levels, published internationally, and now write medical articles focusing on the gut microbiome, mitochondrial health, exercise, mental and cardiovascular wellness, photobiomodulation, and circadian rhythms. He writes at Mike’s Substack.
Cholesterol Synthesis and Biological Necessity
Cholesterol is one of the body’s most tightly regulated and energetically expensive molecules; however, it is still widely portrayed as something to restrict or minimize.
Every nucleated cell can synthesize it, and several organs use substantial metabolic resources for its local production. In some tissues, most notably the brain, dietary cholesterol does not materially influence cholesterol content under normal physiological conditions because lipoprotein-bound cholesterol does not cross the intact blood–brain barrier.
This raises a central biological question: if cholesterol can simply be obtained from food, why did evolution invest in such an elaborate and tightly regulated system to manufacture it internally?
The answer lies in the indispensable structural, metabolic, and signalling roles of cholesterol and the biological necessity of local, on-demand synthesis.
Cholesterol is not only useful but also essential for multicellular life.
This article explores:
de novo cholesterol biosynthesis,
focusing on the liver, astrocytes in the brain, and adrenal glands, and
clarifies the persistent misconception that dietary cholesterol is required for good human health.
Cholesterol: An Essential Structural and Signaling Molecule
Cholesterol is indispensable to mammalian cell membranes, where it governs fluidity, permeability, and the behavior of embedded proteins. By inserting between phospholipids, it stabilizes membranes against extremes of rigidity or excessive leakiness, preserving both structural integrity and dynamic responsiveness across physiological temperatures. This modulation of membrane microviscosity ensures that ion channels, transporters, and receptors operate within optimal biophysical conditions. In its absence, membranes become fragile, more vulnerable to oxidative injury and osmotic stress, and less capable of maintaining mechanical stability.
No alternative lipid fully reproduces cholesterol’s unique amphipathic properties in mammalian cells.
Beyond structural stability, cholesterol enables membrane curvature, vesicle formation, and receptor-mediated endocytosis, integrating membrane architecture with intracellular trafficking and signaling. Given these critical roles, cellular cholesterol levels are tightly controlled. This regulation is orchestrated primarily through the sterol regulatory element–binding protein-2 (SREBP-2) pathway, which synchronizes endogenous synthesis with receptor-mediated uptake.
When cholesterol levels within the endoplasmic reticulum decline, the SREBP-2–SCAP complex relocates to the Golgi apparatus, where proteolytic activation releases the active transcription factor. It then enters the nucleus to upregulate genes encoding HMG-CoA reductase, the rate-limiting enzyme in cholesterol biosynthesis and the LDL receptor, thereby increasing both de novo production and cellular uptake of circulating LDL particles.
Conversely, when intracellular sterol concentrations rise, this pathway is suppressed. Transcription of synthetic and uptake machinery falls, while excess sterols promote ubiquitin-mediated degradation of HMG-CoA reductase, providing rapid post-translational restraint.
Cholesterol synthesis is further linked to cellular energy status through AMP-activated protein kinase (AMPK), which phosphorylates and inhibits HMG-CoA reductase during energy stress.
Together, these integrated feedback systems ensure that cholesterol availability reflects intracellular demand and metabolic state. Homeostasis is governed by cellular sensing mechanisms, underscoring that cholesterol supply is fundamentally an endogenous, tightly regulated process rather than one dependent on dietary intake.
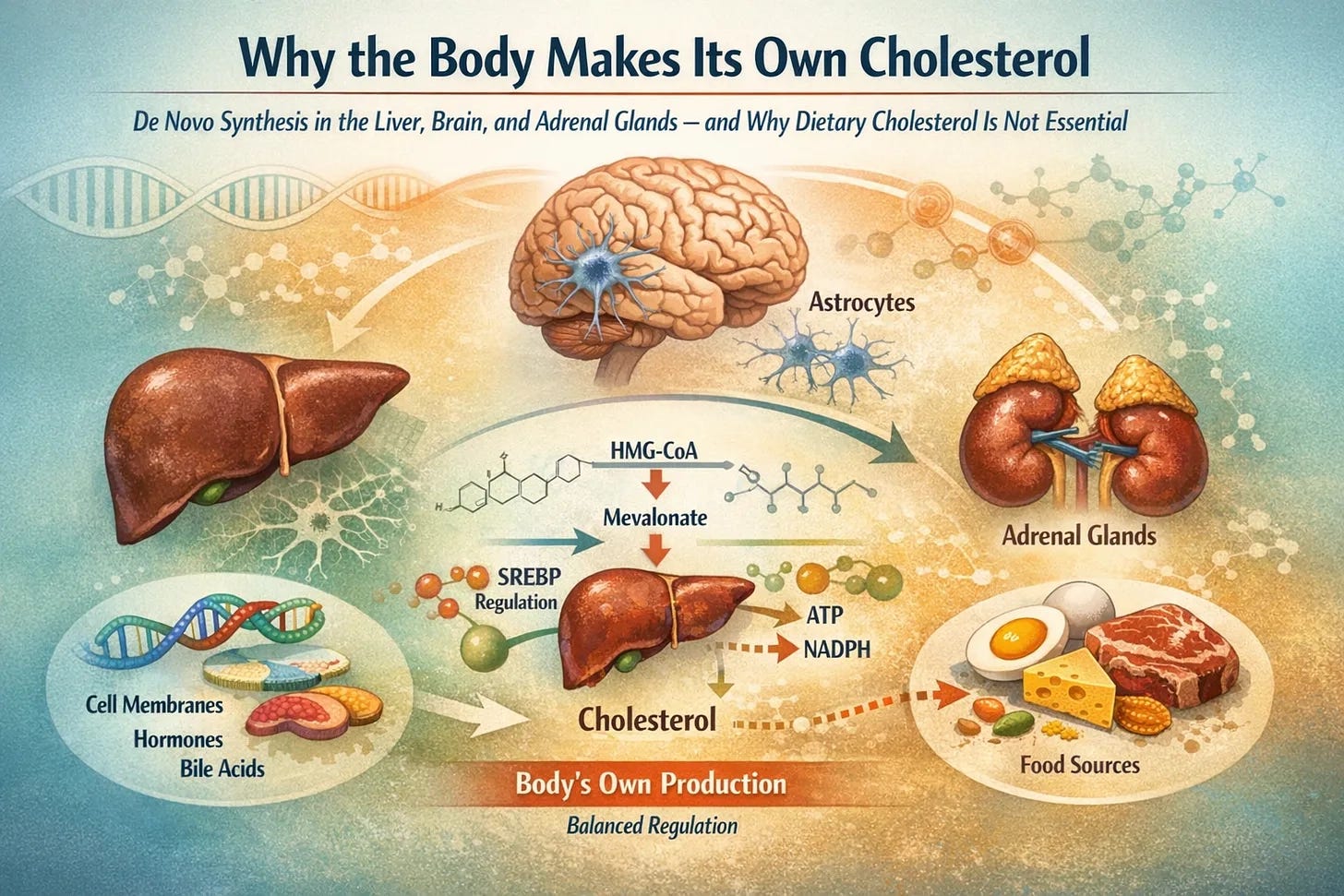
The Liver as the Central Regulator of Whole‑Body Cholesterol Flux
The liver is the single largest source of de novo cholesterol synthesis, producing approximately 700–900 mg/day, with ~50–70% hepatic and the remainder extrahepatic.
It supplies cholesterol for
membranes,
bile acids,
lipoproteins
and coordinates the whole-body distribution via VLDL, LDL, and HDL particles.
Hepatic synthesis responds dynamically to dietary intake; when dietary cholesterol increases, synthesis is downregulated, and when intake decreases, synthesis rises to compensate.
This feedback explains why dietary cholesterol has only modest effects on serum cholesterol levels in most individuals: the liver actively maintains homeostasis rather than passively reflecting dietary intake.
Interindividual Variability in Cholesterol Synthesis and Dietary Responsiveness
Cholesterol synthesis capacity is not uniform across populations.
Twin and family studies suggest that 40–60% of the variation in serum cholesterol levels is genetically determined, reflecting polymorphisms in enzymes of the mevalonate pathway and regulatory proteins involved in intracellular cholesterol sensing. These differences create a spectrum from relatively “high synthesisers” to “low synthesisers.” Two individuals consuming the same diet may exhibit different rates of endogenous production and distinct steady‑state lipid profiles.
This biological diversity underlies the phenomenon of “hyper‑responders” and “non‑responders” to dietary cholesterol intake. Approximately one‑third of individuals show measurable increases in serum cholesterol when dietary cholesterol rises, often due to lower baseline synthesis and higher intestinal absorption efficiency, whereas others maintain relatively stable serum levels because endogenous synthesis adapts or absorption is limited.
Hyper‑responders may derive some benefit from dietary modification, but even in this group, changes in LDL-C may reflect shifts toward larger LDL and higher HDL without clear deterioration in standard risk ratios. Age and metabolic health add further variation: hepatic synthesis and regulatory activity tend to decline with age; however, this trajectory is heterogeneous and influenced by genetics, insulin sensitivity, and cumulative environmental exposures.
Together, these differences reinforce a central principle: cholesterol homeostasis is governed primarily by endogenous regulatory mechanisms, but the efficiency and responsiveness of these mechanisms vary meaningfully between individuals.
Gut Microbiota and Sterol Flux: Modulation Without Substitution
The gut microbiome plays an important but often misunderstood role in cholesterol metabolism.
It does not produce cholesterol for the host and does not determine cholesterol availability at the cellular or tissue level; rather, it influences the amount of cholesterol and bile acids that are absorbed, recycled, or excreted, thereby modulating hepatic demand without replacing endogenous synthesis.
Many gut bacteria express bile salt hydrolases that deconjugate bile acids, thereby reducing their solubility and altering micelle formation. Less efficient bile acid recycling increases faecal sterol loss, depleting the bile acid pool and prompting the liver to upregulate de novo synthesis of bile acids from cholesterol and increase LDL receptor activity to “pull” cholesterol from the circulation. This mechanism helps explain the modest LDL-lowering effects of bile acid sequestrants, certain fibres, and some dietary patterns without altering the fundamental requirement for endogenous production.
Certain microbial species convert cholesterol into coprostanol, a poorly absorbed sterol that is excreted in the stool.
Although this reduces intestinal cholesterol absorption, it triggers compensatory hepatic synthesis, reinforcing reliance on internal production. Microbial fermentation products, particularly short-chain fatty acids (SCFAs), further influence hepatic lipid metabolism by modulating signaling pathways such as AMPK and SREBP‑2. Acetate can serve as a substrate for lipid synthesis, whereas propionate is generally associated with the inhibition of hepatic cholesterol production. These effects fine-tune cholesterol synthesis and lipoprotein output but do not substitute for synthesis itself.
In effect, the microbiome adjusts the accounting, how much the liver must make or recycle without changing the underlying reality that cholesterol is synthesized endogenously, locally, and on demand.
Metabolic and Pathological Modifiers of Endogenous Cholesterol Synthesis
Although endogenous synthesis governs cholesterol homeostasis under normal physiological conditions, certain metabolic and pathological states can modify this balance.
In severe malnutrition, prolonged caloric restriction, sepsis, or acute critical illness, hepatic cholesterol synthesis can be markedly suppressed, and circulating cholesterol levels may fall precipitously, reflecting systemic metabolic stress rather than dietary insufficiency. In such contexts, dietary cholesterol may assume temporary clinical relevance to prevent critical deficiency, but this reflects pathological inhibition of synthesis rather than failure of the normal homeostatic model of cholesterol regulation.
Pharmacological inhibition introduces another nuanced scenario. Statins reduce cholesterol synthesis by inhibiting HMG‑CoA reductase, and tissue exposure varies according to drug characteristics, with lipophilic agents having greater central penetration than hydrophilic agents do. The brain largely maintains autonomous cholesterol synthesis, and most peripheral tissues compensate through feedback mechanisms; however, in theory, profound or prolonged synthesis inhibition in vulnerable individuals could make dietary cholesterol relatively more important in specific tissues than in healthy individuals. Such scenarios are uncommon and context-dependent.
Rare genetic defects in cholesterol biosynthesis represent a clear exception. Individuals with inherited enzyme deficiencies cannot produce sufficient endogenous cholesterol and often require targeted dietary supplementation or pharmacological strategies to partially bypass the metabolic block. These uncommon disorders underscore the broader rule: in healthy physiology, endogenous synthesis is sufficient; when intrinsic synthetic capacity is impaired, dietary provision becomes necessary.
Much of the data supporting the modest impact of dietary cholesterol on serum lipids is derived from Western populations with relatively stable access to animal products. Evidence from non‑Western or historically low animal-product populations remains limited, and direct in vivo measurement of hepatic synthesis in diverse human groups is technically challenging and underutilized, leaving gaps in our understanding.
These limitations do not overturn the central role of endogenous synthesis but clarify the physiological boundaries within which this principle operates.
Brain Cholesterol Metabolism: A Closed and Autonomous System
The adult brain contains ~20–25% of the body’s cholesterol, yet only minimal net cholesterol influx occurs from the circulation under normal conditions, so CNS pools are maintained largely by local synthesis and slow turnover.
Under normal conditions, peripheral lipoprotein‑bound cholesterol has very limited access to the CNS across an intact BBB; therefore, brain cholesterol is largely maintained by local synthesis.
Astrocytes are the principal producers of cholesterol, synthesizing it de novo, predominantly via the Bloch pathway, and supplying it to neurons via ApoE-containing lipoproteins.
Neurons have limited synthetic capacity and rely heavily on astrocytic production for synaptic membranes and signaling; mature synaptogenesis is tightly coupled to astrocyte-derived cholesterol. This isolation creates a distinct metabolic timeline: while circulating cholesterol turns over within days, brain cholesterol has a half-life measured in years, reflecting the brain’s commitment to stability.
The blood–brain barrier shields neural tissue from inflammatory lipoproteins, dietary sterols, and oxidized lipids that can interfere with neurotransmission; therefore, astrocytes effectively function as dedicated lipid factories within this protected environment.
Astrocyte-derived cholesterol acts as both a structural substrate and a neurotrophic factor, supporting synaptogenesis, dendritic expansion, and maintenance of excitatory and inhibitory synapses. When astrocytic cholesterol synthesis is impaired experimentally, neurones retract dendritic spines, synapses are lost, and neurotransmission is disrupted.
Altered cholesterol handling between glia and neurons is increasingly implicated in Alzheimer’s disease and other neurodegenerative conditions.
Excess cholesterol in the brain is converted into oxysterols, notably 24S‑hydroxycholesterol, which can cross the blood–brain barrier for elimination, maintaining equilibrium while preserving brain autonomy.
. https://doi.org/10.1186/s43556-025-00321-3")
The neuronal enzyme CYP46A1 catalyses this conversion and represents the primary route for cholesterol elimination in the brain. Disruption of CYP46A1 impairs cholesterol homeostasis and is linked to neurodegenerative phenotypes in experimental models. Other oxysterols, such as 27‑hydroxycholesterol, traverse the barrier bidirectionally and participate in peripheral–central sterol exchange, providing additional regulatory layers.
Adrenal Cholesterol Utilization and Steroidogenesis Under Stress
The adrenal cortex relies on cholesterol to synthesize cortisol, aldosterone, and adrenal androgens. Under basal conditions, much of this cholesterol is derived from lipoprotein uptake, particularly LDL, and stored cholesteryl esters. During stress or ACTH stimulation, adrenal cells rapidly mobilise stored cholesteryl esters and upregulate local synthesis to sustain steroid output. Steroidogenesis cannot depend on dietary cholesterol arriving “on time”; it requires a constant, regulated supply via stored pools and de novo synthesis to meet the acute and chronic demands of steroid hormones.
Common Clinical Misconceptions About Cholesterol
Despite these mechanistic distinctions, several misunderstandings persist in clinical practice.
First, cholesterol is a molecule, and LDL is a transport particle.
Cholesterol is carried within lipoproteins, such as LDL, HDL, VLDL, and chylomicrons; LDL is not “cholesterol” but a vehicle that contains cholesterol and other lipids. Cardiovascular risk is primarily related to the number of apoB-containing particles (apoB is present in LDL, VLDL, IDL, and lipoprotein(a)), rather than the cholesterol molecule itself. Treating “cholesterol” as a monolithic entity obscures this key distinction between the cargo and carrier.
Second, dietary cholesterol exerts only a modest influence on serum cholesterol levels in most individuals.
Although cholesterol is biologically essential for every cell, it is nutritionally non-essential because endogenous synthesis can meet physiological requirements. Intestinal absorption of dietary cholesterol varies considerably between individuals, and hepatic production is downregulated when intake increases, buffering changes in circulating concentrations. Controlled metabolic ward and feeding studies consistently demonstrate that, in most people, adding dietary cholesterol produces relatively small changes in LDL-C compared with alterations in overall dietary pattern.
Approximately one-third of individuals exhibit greater sensitivity to dietary cholesterol and are often described as “hyper-responders.”
In these individuals, LDL-C and HDL-C commonly rise in parallel, and LDL particles may shift toward larger, less dense subclasses. While LDL-C may increase modestly, the overall change is typically smaller than that produced by saturated fat intake, insulin resistance, systemic inflammation, excess adiposity, or genetic factors such as ApoE genotype. For the majority of individuals, these metabolic determinants exert substantially greater influence on atherogenic lipoprotein levels than dietary cholesterol intake alone.
Implications for Lipid‑Lowering Therapy and Cardiovascular Risk
Understanding endogenous cholesterol synthesis can help reframe the mechanisms by which lipid-lowering therapies reduce cardiovascular risk. Statins primarily lower hepatic cholesterol synthesis, thereby reducing VLDL output and downstream apoB‑containing particle numbers. Their benefit arises not because cholesterol itself is inherently harmful but because excess hepatic particle production increases the atherogenic burden.
At the same time, cholesterol synthesis remains essential for brain function, steroid hormone production, and membrane integrity. Therefore, therapeutic strategies must balance cardiovascular risk reduction with the preservation of these fundamental physiological roles, particularly during long-term or high-intensity treatment.
Accounting for Interindividual Variability in Clinical Practice
Substantial interindividual variability exists in cholesterol synthesis capacity and responsiveness to dietary intakes. Differences in intestinal absorption efficiency and endogenous synthesis underpin the observed spectrum of “hyper‑responders” and “non‑responders” to dietary cholesterol intake.
Hyper‑responders may experience modest LDL-C increases with higher dietary cholesterol intake and can benefit from targeted dietary modifications. However, dietary restriction alone is often insufficient when hepatic overproduction or impaired clearance is predominant. Non-responders typically require attention to metabolic drivers such as insulin resistance, carbohydrate quality, physical inactivity, and excess adiposity rather than cholesterol intake per se.
Targeting Upstream Drivers of Hepatic Lipoprotein Production
Clinical management should prioritize upstream disturbances that influence hepatic lipoprotein output. In insulin-resistant states, improvements in glycemic control, reduction of visceral adiposity, and increased physical activity can reduce hepatic VLDL overproduction. In non-alcoholic fatty liver disease, weight loss and metabolic optimization improve hepatic lipid handling and may lower apoB-particle output. Tailoring macronutrient composition to the metabolic phenotype, rather than uniformly restricting dietary cholesterol, is more likely to meaningfully influence the apoB particle burden and cardiovascular risk.
Situations Requiring Deviation From the “Synthesis Is Sufficient” Model
Deviations from the general framework are appropriate in selected circumstances. In acute critical illness with markedly suppressed protein synthesis, temporary dietary support may be reasonable. Patients with rare biosynthetic defects require specialized care, and individuals receiving potent synthesis inhibitors should be monitored for tissue‑specific effects, including cognitive or endocrine symptoms. In such cases, switching from lipophilic to more hydrophilic statins may be considered if central side effects are suspected. Dietary cholesterol reduction may still play a role in selected high-risk populations, such as those with familial hypercholesterolaemia, but should be framed as one component of a broader strategy targeting apoB-particle overproduction and impaired clearance.
Communicating the Concept
For patients, the explanation can remain simple: most cholesterol in the body is synthesized by the liver and other tissues according to physiological demand, and when dietary intake falls, endogenous production usually increases to compensate. What matters more than restricting dietary cholesterol in isolation is supporting the healthy regulation of hepatic synthesis and lipoprotein metabolism through weight management, physical activity, metabolic control, adequate sleep, and dietary patterns that promote insulin sensitivity.
Conclusion: Biological Independence and Physiological Sufficiency
The body’s capacity to synthesize cholesterol de novo reflects its biological indispensability rather than metabolic redundancy. Evolution has invested heavily in tightly regulated synthetic pathways that ensure cholesterol availability whenever and wherever required, independent of dietary intake. The liver, brain, and adrenal glands maintain distinct, locally regulated synthesis programs, each tuned to tissue-specific demands and insulated, though not entirely isolated from fluctuations in external supply.
Dietary cholesterol is therefore biologically optional.
It can be used, but it is not required to sustain normal physiology in healthy individuals. Cholesterol homeostasis is primarily governed by endogenous synthesis, intracellular sensing, transport, and feedback regulation, with dietary intake exerting only a secondary and highly variable influence. When endogenous synthesis is impaired by pathology, pharmacologic inhibition, or rare genetic defects, dietary provision may assume temporary or supportive relevance; however, these exceptions highlight rather than undermine the central role of intrinsic production. Framing cholesterol metabolism from this perspective clarifies persistent misconceptions in clinical practice.
Cholesterol is an essential molecule, and atherogenic risk is driven by dysregulated lipoprotein particle production and clearance. Effective risk assessment and intervention therefore require attention to hepatic lipoprotein output, metabolic health, and individual regulatory capacity, rather than dietary cholesterol restriction in isolation.
Cholesterol synthesis is not a biological error to be minimized, but a hallmark of physiological self‑sufficiency and a foundational feature of mammalian life.
References
Aderhold, A., & Alexaki, V. I. (2025). Lipid metabolism in the adrenal gland. Frontiers in Endocrinology, 16, Article 1577505.
Cui, D., Yu, X., Guan, Q., Shen, Y., Liao, J., Liu, Y., & Su, Z. (2025). Cholesterol metabolism: Molecular mechanisms, biological functions, diseases, and therapeutic targets. Molecular Biomedicine, 6(1), Article 72.
Fernández-Ruiz, I. (2024). Gut bacteria can break down cholesterol. Nature Reviews Cardiology, 21, 357.
Gomez-Sanchez, C. E., & Gomez-Sanchez, E. P. (2024). Cholesterol availability and adrenal steroidogenesis. Endocrinology, 165(4), Article bqae032.
Li, D., Zhang, J., & Liu, Q. (2022). Brain cell type-specific cholesterol metabolism and implications for learning and memory. Trends in Neurosciences, 45(5), 401–414.
Savulescu-Fiedler, I., Dorobantu-Lungu, L. R., Dragosloveanu, S., Benea, S. N., Dragosloveanu, C. D. M., Caruntu, A., Scheau, A. E., Caruntu, C., & Scheau, C. (2025). The cross-talk between the peripheral and brain cholesterol metabolisms. Current Issues in Molecular Biology, 47(2), Article 115.
Vartiainen, E., Liyanage, D., Mazureac, I., Battaglia, R. A., Tegtmeyer, M., He, J. X., Räsänen, N., Sealock, J., McCarroll, S., Nehme, R., & Pietiläinen, O. (2025). Astrocytic-supplied cholesterol drives synaptic gene expression programs in developing neurons and downstream astrocytic transcriptional programs [Preprint]. bioRxiv.
Yamauchi, Y., Sharpe, L. J., & Brown, A. J. (2026). Balancing cholesterol metabolism in the liver and gut: Perspectives in health and disease. Nature Reviews Gastroenterology & Hepatology. Advance online publication.
Disclaimer This article is not intended to replace professional medical advice. If you have specific health concerns or conditions, consult with a healthcare professional for personalized guidance.
 | A guest post by
|
Appreciate this deep dive – perfect timing now that the updated ACC/AHA dyslipidemia guidelines are out.